This research article from Scientific Reports presents an in-depth investigation into the genetic makeup of the SARS-CoV-2 virus found in patients who died from COVID-19. The study’s main objective was to determine if there were specific, harmful (deleterious) genetic mutations that were commonly found in these fatal cases, regardless of where in the world the patient lived. To achieve this, the researchers analyzed 5,724 complete viral genomes from deceased patients in 123 countries across five continents. By comparing these viral genomes to the original reference strain from Wuhan, they were able to map out the evolutionary changes and identify patterns associated with the most severe outcomes of the disease. This type of research, known as genomic surveillance, is vital for tracking the evolution of the virus.
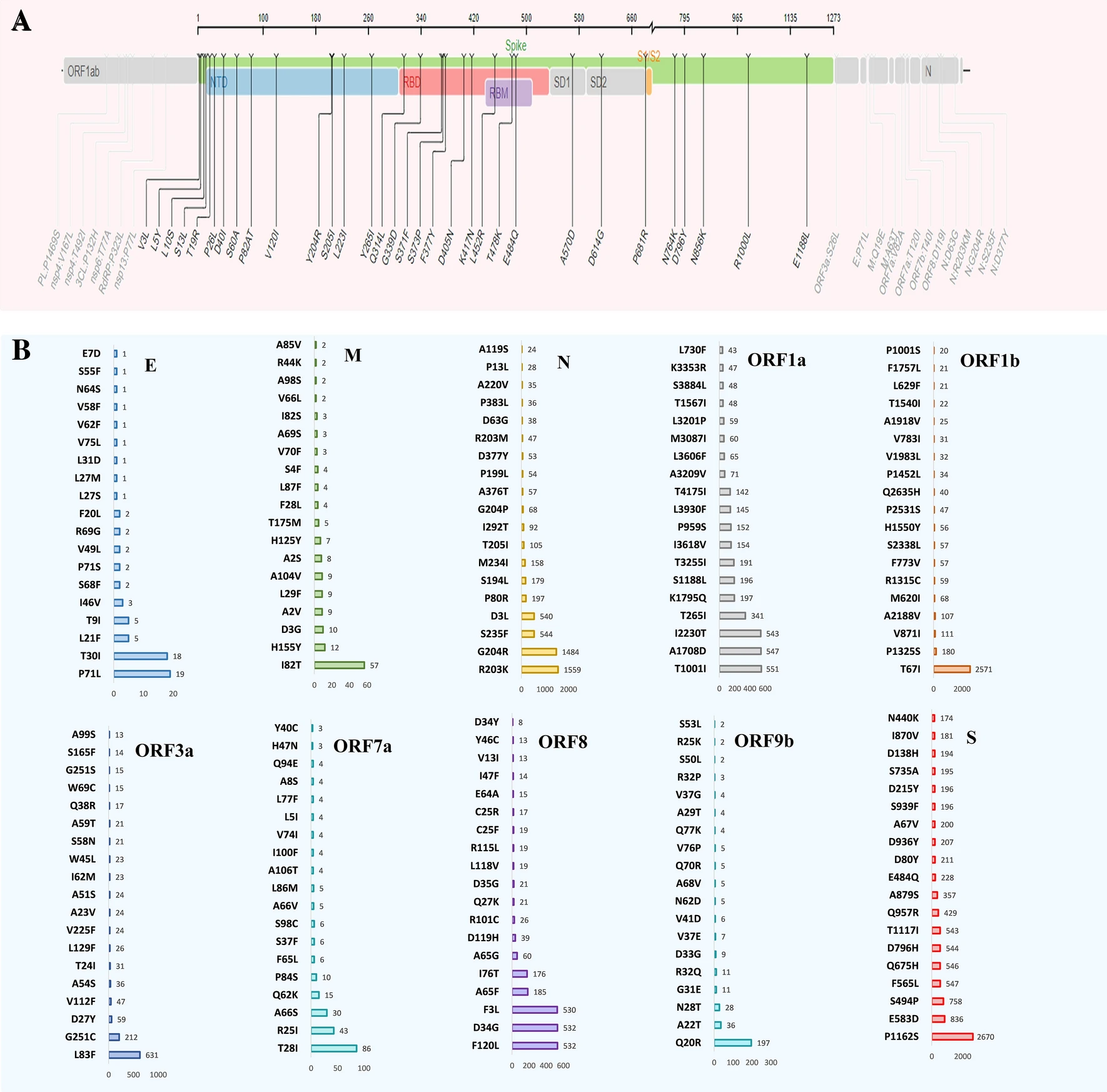
Omicron’s spike protein contained 876 mutations, including 443 predicted to be harmful, with 256 being unique non-synonymous changes; most were concentrated in the receptor-binding domain, which directly interacts with human cells, likely contributing to Omicron’s high transmissibility and ability to evade neutralizing antibodies. In contrast, the Delta variant, analyzed from 1,884 sequences, had 4,468 mutations, 66% unique, with many occurring in amino acid positions 911–924—regions associated with B-cell and T-cell immune recognition—indicating a different evolutionary focus on immune evasion rather than binding efficiency alone. These patterns suggest that while Omicron has evolved primarily to spread more efficiently, Delta’s changes targeted the immune system’s ability to detect and respond to it. The presence of persistent, unique, and harmful mutations in Omicron points to potential targets for updated vaccines or antiviral drug development, as focusing on these mutations could improve disease control and prevention strategies. Our findings emphasize the importance of continuous global genomic surveillance, which enables early detection of significant mutations and guides the timely adaptation of public health measures, vaccines, and treatments. SARS-CoV-2’s rapid and ongoing genetic evolution means that static solutions are insufficient; instead, a proactive approach combining real-time monitoring, advanced computational analysis, and targeted biomedical interventions is essential to staying ahead of the virus and mitigating its health, social, and economic impacts worldwide.
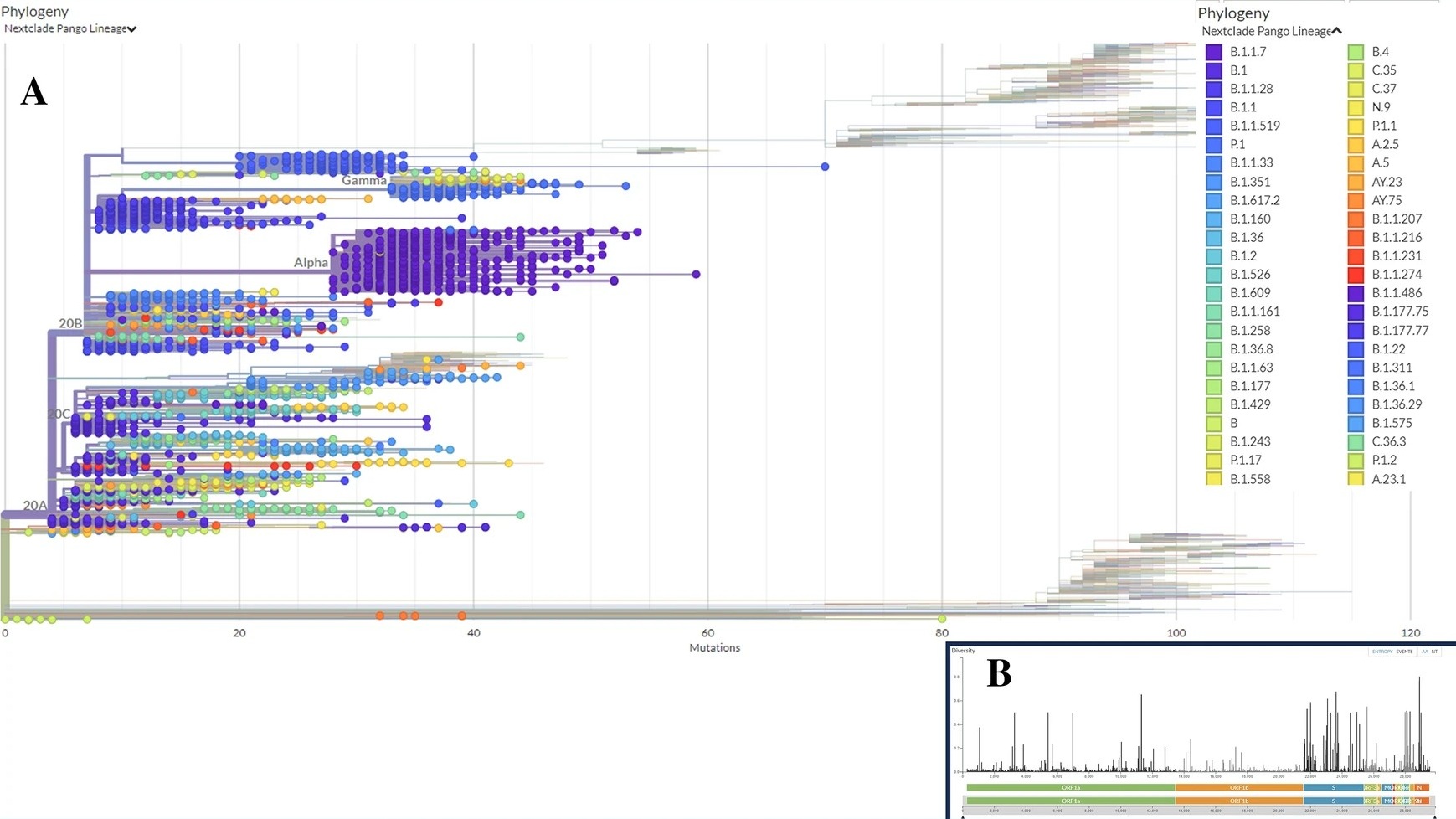
One of the study's key contributions is the detailed mapping of the different variants and clades of SARS-CoV-2 that were prevalent among deceased patients. The analysis revealed that the viral genomes belonged to 21 distinct evolutionary branches, or clades. Notably, the Alpha variant was the most predominant, followed by the Beta and Gamma variants, indicating that these specific variants of concern were significantly associated with mortality during the study period. The research also uncovered interesting geographical patterns; for example, the 'G' clade was most common in fatal cases in Asia, Africa, and North America, whereas the 'GRY' clade was dominant in Europe. This suggests that the virus evolved in slightly different ways as it spread through different global populations. Demographically, the study found that a majority of the sequenced genomes came from male patients, reinforcing findings that males faced a higher risk of severe outcomes. By identifying which viral lineages were most frequently linked to death, this research provides crucial information for public health authorities to prioritize the monitoring of variants that may pose a greater danger.